Remote Asymmetric Induction Using Acetate-Type Vinylketene Silyl N,O‑Acetals【文献紹介】
文献情報
タイトル:Remote Asymmetric Induction Using Acetate-Type Vinylketene Silyl N,O-Acetals
著者:Naoya Sagawa, Haruka Sato, Seijiro Hosokawa* (*Department of Applied Chemistry, Faculty of Science and Engineering, Waseda University)
基本情報:Org. Lett., 2017, 19, 1, 198-201.
受領日:2016/11/22
出版日:2016/12/13
DOI:10.1021/acs.orglett.6b03476
概要
不斉補助基を有する酢酸型ビニルケテンシリル N,O-アセタールを用いたビニロガス向山アルドール反応による遠隔不斉誘導に成功した。クロトン酸およびL-バリンから得られるシリルN,O-アセタールは、SnCl4およびBF3・OEt2で処理することにより、O-シリル化5R-および5S-付加体をそれぞれ選択的に得ることができた。また、SnCl4を介したシリルジエノールエーテルの異性化を見いだし、得られた主異性体は高い反応性を示し、高い立体選択性でγ-付加物を与えることがわかった。
日本語訳
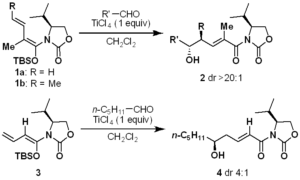 Scheme 1. Previous Results on Remote Asymmetric Induction Using Vinylketene Silyl N,O-Acetals
Scheme 1. Previous Results on Remote Asymmetric Induction Using Vinylketene Silyl N,O-Acetals遠隔不斉誘導反応は、有機化学における挑戦的な課題として研究されてきた1)。開発された系は限られているが、天然物合成の強力なツールとして使われてきた2)。我々は、ポリケチドの短段階合成を実現するための方法論・戦略を研究する過程で、ビニルケテンシリルN,O-アセタール 1a、1b を用いた遠隔不斉誘導反応を開発した (Scheme 1, eq 1)3)。この反応では、不斉中心と多官能基化炭素鎖が同時に導入されるため、ポリプロピオネートの短段階合成を確立することができた。また、1a、1b の結晶は調製が容易であり、安定であることから、親しみやすい化合物である。そのため、この方法論は天然物合成に応用されている4)。さらに、1b を用いた類似の反応を開発し、様々な種類のポリプロピオネートを合成している。2004年の報告では、α-メチル欠損型ジエノラート 3 は、反応部位が不斉補助基から遠く離れ、立体選択性が中程度に低下するジエン鎖を有することも明らかにした (Scheme 1, eq 2)3a)。 また、ジエノールエーテル 3 はカラムクロマトグラフィーによる精製が不安定であることが判明し、そのため、この反応では低収率で生成物 4 が得られてしまう。この反応を改良することで、短段階でポリアセテート骨格を構築する新しい方法論が得られると期待される。本発表では、新たに設計したキラルなジエノラートを用いた改良型ビニロガス向山アルドール反応について紹介する。
1a のX線結晶構造図をFigure 1に示す。オキサゾリジノン環は、ジエン鎖に対してほぼ垂直に配置されている(Figure 1A)。この立体配置は、窒素の不対電子をジエンに近づけないためであり、これが 1a の安定性の理由である。オキサゾリジノン環は、TBSのメチル基とジエノール側鎖に挟まれているため、回転できないように見える。イソプロピル基のメチル基の1つは、四置換オレフィンに重なっている。CDCl3溶液中における 1a の同様の立体配置は、H3とイソプロピル基のメチル基とのNOE相関によって観察された(Figure 1B)。
一方、3 の側鎖の伸長方向(Scheme 1, eq 2)は、1a の側鎖の伸長方向とは異なっていた。反応部位であるジエン 3 の末端炭素は、オキサゾリジノン環上のイソプロピル基から遠いことが望ましい。そこで、イソプロピル基の方向とジエノールエーテルの安定性を改善するために、Figure 2に示す 5 のような新たなビニルケテンN,O-アセタールを設計した。不斉補助基がジエニル基に結合する場合、C5′位の置換基がイソプロピル基をジエンに被せるようにする必要がある。そこで、オキサゾリジノン環のC5′位にジメチル基またはジフェニル基を有するSuperQuats補助基を採用することにした。さらに、TBSの代わりにTBDPSを用いれば、ビニルケテンシリルN,O-アセタールはより安定になる。
イミド 6 のO-シリル化により、5′,5′-ジフェニルオキサゾリジノンとtert-ブチルジフェニルシリル基を有する新しいビニルケテンシリルN,O-アセタール 5 が単一異性体として得られた7b)(Scheme 2)。ジエノールエーテル 5 は、X線結晶構造解析が可能な結晶として得られた(Figure 3)。5 の結晶構造から、オキサゾリジノン環上のフェニル基の1つがイソプロピル基の方向を決定し、別のフェニル基がTBDPS基を押し下げていることが明らかになりました。したがって、TBDPSのフェニル基の1つは、ジエン鎖の下に向いていた。オキサゾリジノンのイソプロピル基は、ジエンの末端炭素、つまり求電子剤との反応部位から遠い位置にあった。CDCl3中でH4′とTBDPSのtert-ブチル基の間にNOE相関が観測されたことから、この立体配置は溶液中でも可能であると考えられる(Scheme 2)。
ビニルケテンシリルN,O-アセタール 5 を入手し、種々のルイス酸を用いたベンズアルデヒドとのビニロガス向山アルドール反応を行った (Table 1)。TiCl4を用いて5S-isomer 8 を主生成物として収率および立体選択性よく得た (Table 1, entry 1)。ジエノールエーテル 3 (Table 1, eq 2) と比較して,収率および立体選択性が向上しており,これはキラルなビニルケテンシリルN,O-アセタールの改質が反映されている。また、生成物には TBDPS基が付加しており、保護されたアルドール付加体が1ステップで得られた。BF3・OEt2では、同程度の収率と選択性で主生成物が得られ(entry 2)、TMSOTfでは低収率で生成物が得られた(entry 3)。SnCl4を使用すると、最も優れた収率と立体選択性が得られた (entry 4)。この反応では、8 とはC5位が逆の配置の異性体である5R-異性体 7 が主生成物であった。5 の5’-非置換誘導体および5’,5’-ジメチル誘導体を用いると収率と立体選択性が低下した(entry 5, 6)8)。そこで、5’,5’-ジフェニルオキサゾリジノン誘導体 5 を採用し、さらに研究を進めた。
次に,SnCl4存在下での各種アルデヒドとの反応を検討した(Table 2)。p-ブロモおよび p-メトキシベンズアルデヒドも,優れた選択性で高収率にO-保護付加体を与えた(Table 2, entry 2, 3)。p-ニトロベンズアルデヒド (entry 4) の場合、反応はゆっくりと進行し、O-保護付加体は低収率で得られたが、選択性は良好であった。遊離のヒドロキシ基を有する生成物は,ジアステレオマーの混合物として得られた。直鎖および分枝側鎖を含む飽和アルキルアルデヒドは,O-保護付加体を高い立体選択性で収率よく得た (entry 5-7).α,β-不飽和アルデヒドの場合 (entry 8, 9) 、反応は良好な収率で、良好から中程度の選択性でO-保護付加体を与えた。ジエノールエーテル 5 とチグリックアルデヒドの混合物を BF3・OEt2 で処理すると、立体選択性が切り替わり、5S-異性体 8 が優れた立体選択性をもって高収率で得られた (entry 10)。
SnCl4を用いた立体選択性の切り替えに興味を持った我々は、アルデヒドを用いない対照実験を行った。ビニルケテンシリルN,O-アセタール 5 をジクロロメタン中 -78 ℃で SnCl4と処理すると、E異性体 9 が主異性体である幾何異性体の混合物がスムーズに得られた。トルエンを用いて,この混合物を同等の立体選択性で定量的に得た。得られたビニルケテンシリルN,O-アセタール 5 および 9 の混合物を,同じ条件でベンズアルデヒドおよび SnCl4との反応に供したところ,Table 1 のentry 4 と同じ結果が得られた。一方,5 と 9 の混合物を BF3・OEt2の存在下でベンズアルデヒドと処理すると,主生成物として 7 が得られた。BF3・OEt2はビニルケテンシリルN,O-アセタール 5 の異性化を促進しないため、本反応の主生成物はScheme 3のE異性体 9 由来となった。
BF3・OEt2 を用いてわずか1分で反応を進行させると,E-エノラート 9 は消失し,Z-エノラート 5 は反応混合物に残存した (Scheme 4)。このことから,E-エノラート 9 は Z-エノラート 5 よりも反応性が高く,高い立体選択性で 7 に変換されたことがわかった(dr 15:1)。
次に,末端メチル基を有するビニルケテンシリルN,O-アセタール 10 を用いて,ビニロガス向山アルドール反応について検討した(Table 3)。5′,5′-ジメチルオキサゾリジノン誘導体 10m と5′,5′-ジフェニルオキサゾリドン誘導体 10p の収率と反応の立体選択性を比較検討した。また、銅(II)トリフラートの添加により、反応の再現性が向上した11)。 5′,5′-ジメチルオキサゾリジノン誘導体 10m は、収率および立体選択性に優れたアンチ付加体 11 を得た(Table 3、entry 1)。5′,5′-ジフェニルオキサゾリジノン誘導体 10p も収率および立体選択性を低下させて 11 を与えた(entry 2)。これらの誘導体の違いは、プロピオンアルデヒドに対する反応性において明確に見出された(entry 3, 4)。10m は良好な収率と立体選択性を保持していたが(entry 3)、10p は中程度の収率と選択性を与えた(entry 4)。したがって、5′,5′-ジメチルオキサゾリジノン誘導体 10m は、ジフェニル誘導体 10p よりも好ましいものであった。
10m を用いたビニロガス向山アルドール反応を様々なアルデヒドで行った(Table 4)12)。ベンズアルデヒド、p-ブロモベンズアルデヒド、p-ニトロベンズアルデヒドなどの芳香族化合物は、高い立体選択性で O-シリル化アンチ付加体 11 を良好な収率で与えた (Table 4, entry 1-3)。プロピオンアルデヒドは高い選択性でTBDPS保護された付加体 11 を与え、イソブチルアルデヒドおよびシクロヘキサンカルボキシアルデヒドを含む分枝飽和アルデヒドは良好から中程度の選択性でアンチ付加物を与えた(entry 4-6)。しかし、α,β-不飽和アルデヒドであるチグリックアルデヒドは共役付加を促進し、ジアステレオマー混合物としてマイケル付加体を与え、アルドール付加体を与えなかった(entry 7)。
これらの結果は、ビニルケテンシリルN,O-アセタール 5 のX線結晶構造解析、Sn(IV)を介したジオメトリーシャッフリング、ビニロガス向山アルドール反応など、Scheme 5 に示す反応機構を示唆するものであった。5 のX線結晶構造解析から、TBDPSのフェニル基の1つがジエンの下面を覆っていることがわかった。したがって、求電子剤が上面から接近するはずである。さらに、優先遷移状態におけるH2の立体反発を避けるために,アルデヒドを配置した。これらの要因から、Scheme 5に示すような立体制御モードが導かれると考えられる。TiCl4と BF3・OEt2を用いた場合、遷移状態 12 を経由して反応が進行し、付加体 8 が主生成物として得られた(Scheme 5, route A)。一方、SnCl4を用いると 5 は異性化してより反応性の高いジエン 9 となり、上面からの求電子攻撃も可能になった。この面選択性はメチル付加ジエン 10 を用いて証明され、4R-付加体 11 が得られた(Table 3、Table 4)。したがって、この反応の遷移状態は 13 と描かれるかもしれない(Scheme 5, route B)。TBDPS が移動し、アルドール付加体とともに安定なTBDPS-O結合が形成される。
結論として、クロトン酸由来のビニルケテンシリル N,O-アセタールを用いた遠隔不斉誘導反応を開発した。SnCl4がシリルジエノールエーテルの形状を異性化することがわかり、得られた E-エノラート 9 は高い反応性を示し、高い立体選択性で付加体を与えることができた。これまでのScheme 1の反応が 1a および 1b の下面から進行するのに対し、5 および 9 の上面からアルデヒドが接近した。これらの結果は、遠隔不斉誘導の新しいシステムを示唆するものであった。また、ビニルケテンシリル N,O-アセタール 10m は、良好から高い立体選択性で反付加体を与え、ジエノレートの面選択性が示された。これらの反応は、ポリケチドを合成するための容易な方法であると思われる。
注釈
1) 遠隔不斉誘導反応の研究事例 (a) ラジカルリレー機構によるステロイドの遠隔官能基化 (b) 遠隔不斉誘導.環状ヒドロホウ化反応による非環状ジオールへの立体選択的アプローチ (c) スルホキシドを用いた非環状系における遠隔オレフィンの分子内ヒドロキシル化反応 (d) 1,7-不斉誘導によるケトボロン酸還元 (e) (f) 炭素鎖に沿った情報伝達による超遠隔立体構造制御 (g) 1,60以上の不斉誘導によるフォールダマーを介した遠隔立体制御
2) 遠隔不斉誘導反応の応用例 (a) 新たな遠隔不斉誘導を用いた(+)-pedamideの全合成 (b) メチルケトンアルドール付加反応における1,5-不斉誘導 (c) (ジエン)鉄トリカルボニル錯体の1,3-移動反応を利用した遠隔不斉誘導 (d) 抗真菌性マクロライド系抗生物質(+)-Roxaticinの合成 (e) 初の再編成されたネオクレロダンジテルペノイドの合成。(-)-Teubrevin Gへの効率的なアクセスを目指した完全位置選択的三置換フラン環化および中環アルキル化法の開発 (f) 遠隔不斉誘導による(-)-Galanthamineの全合成 (g)
3) 同研究室における過去の遠隔不斉誘導反応の研究事例 (a) キラルなビニルケテンシリルN,O-アセタールを用いたTiCl4による高位置選択的および高立体選択的なビニロガス向山アルドール反応の開発 (b) 天然物の全合成における、ビニルケテンN,O-アセタールを用いた不斉ビニロガス向山アルドール反応 (c) エノラート化学の開発と天然生理活性物質の全合成
参考文献
1) (a) Breslow, R.; Corcoran, R. J.; Snider, B. B., J. Am. Chem. Soc., 1974, 96, 6791. (b) Still, W. C.; Darst, K. P., J. Am. Chem. Soc., 1980, 102, 7385. (c) Hauser, F. M.; Ellenberger, S. R., J. Am. Chem. Soc., 1984, 106, 2458. (d) Molander, G. A.; Bobbitt, K. L., J. Am. Chem. Soc., 1993, 115, 7517. (e) Linnane, L. P.; Magnus, N.; Magnus, P. Nature, 1997, 385, 799. (f) Clayden, J.; Lund, A.; Vallverdu, L.; Helliwell, M. ́ Nature, 2004, 431, 966. (g) Byrne, L.; Sola, J.; Boddaert, T.; Marcelli, T.; Adams, R. W.; ̀Morris, G. A.; Clayden, J., Angew. Chem., Int. Ed., 2014, 53, 151.
2) (a) Yanagiya, M.; Matsuda, F.; Hasegawa, K.; Matsumoto, T., Tetrahedron Lett., 1982, 23, 4039. (b) Evans, D. A.; Coleman, P. J.; Côte,́ B. J. Org. Chem., 1997, 62, 788. (c) Takemoto, Y.; Ishii, K.; Honda, A.; Okamoto, K.; Yanada, R.; Ibuka, T. Chem. Commun., 2000, 1445-1446. (d) Evans, D. A.; Connell, B. T., J. Am. Chem. Soc., 2003, 125, 10899. (e) Efremov, I.; Paquette, L. A., J. Am. Chem. Soc., 2000, 122, 9324. (f) Kodama, S.; Hamashima, Y.; Nishide, K.; Node, M., Angew. Chem., Int. Ed., 2004, 43, 2659. (g) Dias, L. C.; Vieira, A. S.; Barreiro, E., J. Org. Biomol. Chem., 2016, 14, 2291.
3) (a) Shirokawa, S.; Kamiyama, M.; Nakamura, T.; Okada, M.;
Nakazaki, A.; Hosokawa, S.; Kobayashi, S., J. Am. Chem. Soc., 2004, 126,
13604. [文献紹介] (b) Hosokawa, S.; Tatsuta, K., Mini-Rev. Org. Chem., 2008, 5, 1. (c) Hosokawa, S., Yuki Gosei Kagaku Kyokaishi, 2009, 67, 24.
4) (a) Nakamura, T.; Shirokawa, S.; Hosokawa, S.; Nakazaki, A.; Kobayashi, S., Org. Lett., 2006, 8, 677. (b) Jiang, X.; Liu, B.; Lebreton, S.; De Brabander, J. K., J. Am. Chem. Soc., 2007, 129, 6386. (c) Nicolaou, K. C.; Guduru, R.; Sun, Y.-P.; Banerji, B.; Chen, D. Y.-K., Angew. Chem., Int. Ed., 2007, 46, 5896. (d) Nicolaou, K. C.; Sun, Y.-P.; Guduru, R.; Banerji, B.; Chen, D. Y.-K., J. Am. Chem. Soc., 2008, 130, 3633. (e) Lipshutz, B.; Amorelli, B., J. Am. Chem. Soc., 2009, 131, 1396. (f) Paterson, I.; Kan, S. B. J.; Gibson, J. Org. Lett., 2010, 12, 3724. (g) Hosokawa, S.; Matsushita, K.; Tokimatsu, S.; Toriumi, T.; Suzuki, Y.; Tatsuta, K., Tetrahedron Lett., 2010, 51, 5532. (h) Matsui, R.; Seto, K.; Sato, Y.; Suzuki, T.; Nakazaki, A.; Kobayashi, S. Angew. Chem., Int. Ed., 2011, 50, 680. (i) Fujita, K.; Matsui, R.; Suzuki, T.; Kobayashi, S., Angew. Chem., Int. Ed., 2012, 51, 7271. (j) Hartmann, O.; Kalesse, M., Angew. Chem., Int. Ed., 2014, 53, 7335. (k) Miyatake-Ondozabal, H.; Kaufmann, E.; Gademann, K., Angew. Chem., Int. Ed., 2015, 54, 1933. (l) Liao, L.; Zhou, J.; Xu, Z.; Ye, T., Angew. Chem., Int. Ed., 2016, 55, 13263.
5) (a) Mukaeda, Y.; Kato, T.; Hosokawa S., Org. Lett., 2012, 14, 5298. [文献紹介] (b) Tsukada, H.; Mukaeda, Y.; Hosokawa, S., Org. Lett., 2013, 15, 678. (c) Takahashi, Y.; Otsuka, M.; Harachi, M.; Mukaeda, Y.; Hosokawa, S., Org. Lett., 2014, 16, 4106. (d) Nakamura, T.; Kubota, K.; Ieki, T.; Hosokawa, S., Org. Lett., 2016, 18, 132.
6) (a) Davies, S. G.; Sanganee, H. J. Tetrahedron: Asymmetry, 1995, 6,
671. (b) Bull, S. G.; Davies, S. G.; Jones, S.; Polywka, M. E. C.; Prasad, R.
S.; Sangance, H. J. Synlett, 1998, 1998, 519. (c) Bull, S. D.; Davies, S. G.;
Jones, S.; Sanganee, H. J. J. Chem. Soc., Perkin Trans. 1 1999, 387.
7) (a) Gibson, C. L.; Gillon, K.; Cook, S., Tetrahedron Lett., 1998, 39,
6733. (b) Hintermann, T.; Seebach, D. Helv. Chim. Acta, 1998, 81, 2093.
8) Relation of stability and stereoselectivity of vinylketene silyl N,O-acetal was revealed in the catalytic enantioselective vinylogous Mukaiyama aldol reaction: Denmark, S. E.; Heemstra, J. R., Jr., J. Org. Chem., 2007, 72, 5668.
9) (a) Saigo, K.; Osaki, M.; Mukaiyama, T., Chem. Lett., 1975, 4, 989.(b) Carreira, E. M.; Singer, R. A., Tetrahedron Lett., 1994, 35, 4323. (c) Christmann, M.; Kalesse, M. Tetrahedron Lett., 2001, 42, 1269. (d) Boxer, M. B.; Yamamoto, H., J. Am. Chem. Soc., 2006, 128, 48.
10) Although isomerization of ketene silyl N,O-acetal has not been reported, the transformation of silyl enol ether to α-stannylketone was revealed: (a) Nakamura, E.; Kuwajima, I., Chem. Lett., 1983, 12, 59. (b) Gennari, C.; Bernardi, A.; Poli, G.; Scolastico, C., Tetrahedron Lett., 1985, 26, 2373.
11) (a) Krü ger, J.; Carreira, E. M., J. Am. Chem. Soc., 1998, 120, 837. (b) Bluet, G.; Bazan-Tejeda, B.; Campagne, J.-M. ́ Org. Lett., 2001, 3, 3807. (c) Moreau, X.; Bazan-Tejeda, B.; Campagne, J.-M. ́ J. Am. Chem. Soc., 2005, 127, 7288. (d) Bazan-Tejeda, B.; Bluet, G.; Broustal, G.; ́ Campagne, J.-M. Chem. – Eur. J., 2006, 12, 8358. Cu enolate as the intermediate was revealed: Pagenkopf, B. L.; Krü ger, J.; Stojanovic, A.; Carreira, E. M., Angew. Chem., Int. Ed., 1998, 37, 3124.
12) For the case of TiCl4 and BF3·OEt2 employed as the Lewis acid, see the Supporting Information.








