(+)-Mutilinの全合成【文献紹介】
文献情報
| タイトル | Total Synthesis of (+)-Mutilin: A Transannular [2+2] Cycloaddition/Fragmentation Approach |
|---|---|
| 著者 | Han Chen, Zesheng Li, Peng Shao, Haosen Yuan, Si-Cong Chen, and Tuoping Luo |
| 基本情報 | J. Am. Chem. Soc., 2022, 144, 34, 15462–15467. |
| 受領日/出版日 | 2022-07-01/2022-08-22 |
| DOI | 10.1021/jacs.2c06934 |
概要
ジテルペノイド(+)-Mutilinの新規かつエナンチオ選択的な全合成について説明する。6,9-バイシクルを構築するためのクライゼン転位アプローチに続いて、トランスアニュラー [2+2] 光環化付加とそれに続く開環反応が、特徴的な5-6-8プロペラン様骨格を形成するために行われた。その後、後段のアルキル化および還元反応により、合成が完了した。
単離
| 単離 | Pleurotus mutilus (Clitopilus scyphoides) Pleurotus passeckerianus (Clitopilus passeckerianus) |
|---|---|
| 生物活性 | Klebsiella pmeumoniae: MIC = 0.5 μL mL-1 Staphylococcus aureus: MIC = 0.5 μL mL-1 |
| 構造決定 | 1H NMR, 13C NMR, MS, UV, IR, X-ray, ORD curve, Chemical degradation and derivatization |
| 構造的特徴 | 三環式ジテルペン |
誘導体
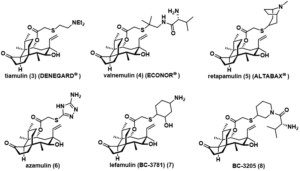
pleuromutilinの誘導体は、医薬品として盛んに研究されている。
tiamulin、valnemulin、retapamulinは既に製品化され、医薬品として使用されている。
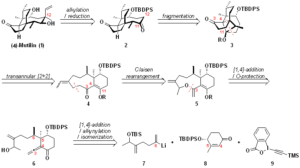
逆合成解析
全文和訳
抗生物質耐性菌による人類の健康への脅威は、新しい抗生物質の発見だけでなく、既存の抗生物質を改良する実用的な方法にも大きな圧力をかけている1)。臨床で使用される天然物抗生物質の重要性は言い尽くせないが、その構造の複雑さゆえに、新規アナログの生成において半合成のアプローチに限界がある2)。デノボ全合成は、効率的に実行されれば、それらを改変する信頼性の高い柔軟な方法を提供する。これは、テトラサイクリン、マクロライド、ストレプトグラミン、アリロマイシン、バンコマイシンに関する研究によって優雅に説明されている3-7)。このシナリオは、このジテルペノイド系抗生物質の合成上の課題に取り組む最近の進歩から、Pleuromutilin(Figure 1, 1)にも拡張されるであろう8, 9)。脱アシル化誘導体 (+)-Mutilin (2) と相互変換可能な (+)-Pleuromutilinは、5-6-8の高密度な官能基を持つ炭素環で、何十年も合成化学者の探究心を刺激し続けている10)。現在までに報告されている6つの合成(Figure S1)のうち、最初のエナンチオ選択的アプローチはProcterと共同研究者により達成され9d)、Herzon 9e) とReisman 9g) の研究グループによる新しい戦略は合成効率を著しく改善し、簡潔な方法で (+)-1 を達成した。最近では、Proninのグループが、外部選択的なDiels-Alder反応、MHATによるラジカル環化、酸化的な環拡張を利用して、1 および 2 への効率的かつ立体制御的なアプローチを報告している9h)。また、Sorensenと共同研究者は、pleuromutilinクラスの抗生物質の簡略化したアナログが抗菌活性を保持していることを明らかにし、このユニークな5,6,8-三環式足場の価値を再確認していることは注目すべきことである11)。
既存の半合成や全合成では到達困難な位置で標的分子を修飾することができるため、1 に代わる戦略はこの分野に有用な付加価値を提供する。ここでは、ユニークなトランスアニュラー[2+2]光付加/フラグメンテーション戦略に基づく (+)-2 の全合成を説明する12)。我々の逆合成分析は、C12(pleuromutilinの番号、全体)の2つの置換基は、対応するエノラートのアルキル化によって中間体 3 に導入できるという仮説から始まった。成功すれば、有機金属試薬の添加9a-d)や置換基を組み込んだビルディングブロック9e-g)を用いたこれまでの戦略とは異なり、合成の後期段階で簡便かつ柔軟にC12を機能化できることになる。3 におけるC11とC4の接続により、C4-C11結合がレトロアルドールフラグメンテーションにより容易に切断される、歪んだシクロブタン系を持つ中間体 4 が得られた13)。同様の中間体が提案されていたが、これまで実現されていないことが指摘された (Figure S2) 14)。4 を構築するための経穴的アプローチでは、エノン 5 を調製する必要がある。高度に置換された9員環の鍛造が困難であることから、我々は型破りなアプローチに踏み切った。その結果、6 は分子内オキシマイケル付加とO-プロテクションにより、アレニルケトン 7 まで遡ることができた。我々は、エノン 9 にビニルリチウム 8 をジアステレオ選択的に共役付加し、アルキニル化(TMS-EBX16)などのシンソン 10 を使用)して異性化すれば、7 と同様に容易に合成できると考えていた。この戦略的な青写真は一見簡単そうに見えるが、変換順序や立体制御の問題を微調整するために、かなりの量の実験的探求が予想された。
順方向(Scheme 1)では、まずTESで保護されたメチル (R)-(+)-ラクテート (11) 17)を、過剰のトリメチルシリルメチルセリウム試薬との反応と硫酸での処理によりアリルシラン 12 に変換した18)。12 の水酸基を脱プロトン化した後、2,3-dibromoprop-1-ene の存在下で HMPA をトリガーとする Brook 転位を行い19)、分離した付加体を TBS で保護して臭化 13 を生成した。その後、エナンチオに富化したエノン 9(調製法については、Supporting Information(SI)を参照)20)に、13 から誘導された有機カップリング試薬の共役付加を行い、エノールエーテル 14 を得、これをTBAFの存在下でTMS-EBX16)と反応させてからシリカゲルで処理すると、2段階で65%の収率でアレニルケトン 15 になった。ジアステレオ選択性の高い共役付加は、C14の嵩高いOTBDPS置換基により、C5四級炭素中心の配置が確保され、制御されていた。TEA-3HFによるTBS基の選択的除去後、分子内オキシマイケル付加を行うための様々な条件を検討した結果(表S1)、16 をtBuOKの還流溶液にtBuOHでゆっくり加えるのが最適で、グラムスケールでエノン 17 を62%の収率で得ることができた。17 のX線構造を調べたところ、このコンフォメーションにおけるA(1,3)歪みを最小化するためには、C10立体中心が重要であることが示唆された21)。エノン 17 を LiHMDS で選択的にγ-脱プロトン化し、Mander 試薬で C-アシル化すると、α-ピロン 18 が得られた22)。主要な副生成物である 19 は、C-アシル化の位置異性体であり、エノン 18 に戻すことができた(SI 参照)。
クライゼン転位はトルエン中でスムーズに進行し、NOESY スペクトル(SI 参照)により確立された C9-C10 アルケンの E 配置を持つ 20 を、おそらくボート状の遷移状態 A を介して得た (Scheme 2)。次に、反応混合物を室温で365 nmのLED(5W)で照射したところ、18 から渡環[2+2]光環化付加生成物 21 が単一のジアステレオマーとして69%の収率で得られた。残念ながら、C10立体中心の配置は、天然物とは逆であった。21 のラクトンを加水分解し、レトロアルドール反応(中間体C)と脱炭酸を行うと、ジケトン 22a が得られ、その構造はX線結晶構造解析により明確に決定された。たとえ間違ったC10立体中心が後工程でエピマー化しても23)、[2+2]光環化付加反応によって正しい立体中心を生成させることに傾倒していた。三重項ビラジカル中間体Bの生成に続く変換がC10の立体化学を決定し24)、基質構造を変更することで微調整が可能であると考えた。そこで、標的分子に存在しないラクトンカルボニル基を除去することにした。その結果、Claisen転位生成物 20 の加水分解と脱炭酸を行い、18 からワンポットで収率70%の 23 が得られた。NMRスペクトルから、室温では 23 のエノール型が優勢であることが明らかであり(SI参照)、その後、容易にシリルエノールエーテル 24 に変換された。24 に放射線を照射すると、3つの主要な生成物、25a/b および 26 が分離不可能な混合物として得られました。一方、25a と 26 は、1:2.2の比率で35%の収率で単離され、26 の脱シリル化生成物であるジケトン 27(6%)と一緒に少量の脱シリル化生成物を得ることができた。25b の脱シリル化が 25a に比べて速いのは、遷移状態においてC17-メチル基と嵩高いTBS保護基の間の歪みがなくなったためと考えられる(紫色の両頭の矢印部分)。渡環[2+2]光付加基質を 20 から 24 に切り替えると、C10立体中心の配置が少なくとも部分的に逆転することから、反応機構を明らかにするためにDFT計算をレトロスペクティブに行った(SIを参照)。簡単に説明すると、20 から生成した中間体Bに関しては、ラクトン部分の制約により、C11-C10結合の形成がC4-C9結合に先行している。一方、24 から生成した三重項ビラジカル中間体Dは、C4-C9結合を先に形成することを好み、C9-C10結合の回転が系間交差やビラジカル再結合と競合して 25a および 25b が得られる中間体Eを得る。予備的ではあるが、この理論的根拠は我々の実験結果とよく一致しており、この興味深いプロセスの詳細を明らかにするためにさらなる研究が必要である。
したがって、渡環[2+2]光環化付加反応 (approach 1) の望ましいジアステレオ選択性は、ビラジカルEの分子内1,5-水素原子移動 (approach 2) が競合して 26 を生成することによって損なわれた。この競合経路を最小化するために、我々は重水素の運動学的同位体効果を利用して不要な副反応を抑制するエレガントな作品からヒントを得た25)。そこで、MeOD/D2O を溶媒としてクライゼン転位生成物 20 の加水分解を行い、18 からグラムスケールで 64%の収率で C2,C12 重水素化 d-23 を得て (Scheme 3)、これをさらに d-24 に転化させる。d-24 の渡環[2+2]環化付加反応により、生成物の比率が変化し、目的のd-22b の形成に有利となり、ワンポットのTBS-脱保護とフラグメンテーションにより47%の収率で単離された。Scheme 2 で定義された機構論的提案と一致し、このシナリオでは、副生成物 26 が単重合 C10 メチレンで形成された(SI を参照)。C6-C16アルケンの顔面選択的水素化によりC6立体化学中心を確保した後、過剰量のLiHMDSを使用してジエノラート中間体を生成した。TIPSOTf当量比を慎重に制御することにより、シクロペンタノン部分の選択的保護が実現し、その後MeIで処理してC12にメチル基が導入された。NOESYデータ(SI参照)により立体化学が確立されたケトン 28 は、エノラートF(X = H)の上面から求電子剤が有利に攻撃されることを意味し、単一のジアステレオマーとして59%の収率で得られた。C12でのジアステレオ選択的なアルキル化の根拠は、C16とC14の間のシンペンタン相互作用を最小化することにより、嵩高い-OTBDPSが擬軸位置になり、C17-メチル基とともにFの下面からの求電子物質のアクセスを遮断することにある。(1) C12 重水素を LDA で除去してエノラート F (X = Me) を生成し、アセトアルデヒドの添加により、Zimmerman-Traxler モデルに沿った単一ジアステレオマーとしてアルドール生成物 29 が収率 49%で得られた; (2) Martin スルフラン脱水により、30 が 84%で得られた、という2段階で残りのビニル基を設置した。最終段階はTIPSとTBDPSの保護基の除去で、シクロオクタンジオンモチーフのレトロマイケルフラグメンテーションが懸念された14a, 27)。プレウロムチロンのメタノール還流下でのアルカリ加水分解に成功したことを励みに27a,28)、30 を THF 還流下で TBAF で処理すると脱保護だけでなく C2 重水素が水素と交換されて (-)-mutilone (31) になることがわかった28)。既報に触発され9f, h)、C11 ケトンの位置およびジアステレオ選択的還元にエタノール中のナトリウムを適用し、(+)-mutilin (2) の合成を達成した。合成した試料 2 のNMRスペクトルデータは、すべて文献で報告されているものと一致した (Table S2)9h)。
以上のことから、我々は、(R)-乳酸メチルとエノン 9 から出発する (+)-ムチリン (2) の全合成を、最長の直列 18工程で達成した(Scheme S1)。エノンの1,4-付加/ビニリデン化、分子内オキシマイケル付加、クライゼン転位に基づく、置換シクロノナノンの構築というユニークな戦略を開発した。また、他の方法では困難な多環式構造へのアクセスを渡環環化付加反応によって実現したことも特徴であり、他の複雑な天然物合成への応用が期待される。さらに、C11ケトン中間体を経由してC12第四級中心を立体選択的に構築した。これは、新しいプレウロムチリン類似体の半合成的調製に役立つと考えられ、この重要な抗生物質の研究領域を拡大する。








