

アメリカのペンシルバニア大学教授であるAmos B. Smith, IIIがNeaumycin Bの全合成を報告した。彼は、これまでに90以上の天然物や生物活性物質を合成している。アニオンリレーケミストリー(ARC)を用いた全合成や抗体を触媒として用いたペプチド合成などが有名。

文献情報
| タイトル | Total Synthesis of-the Reported Structure of Neaumycin B |
|---|---|
| 著者 | Jiaming Ding; Amos B. Smith, III |
| 基本情報 | J. Am. Chem. Soc., 2023, 145, 18240–18246. |
| 受領日/出版日 | 2023-06-22/2023-08-10 |
| DOI | 10.1021/jacs.3c06573 |
概要
総収率2.3%で、報告されている構造のNeaumycin Bを立体選択的に合成した。鍵反応として、ニッケル触媒を用いた還元的クロスカップリング/スピロケタリゼーションが挙げられる。立体構造はX線結晶構造解析を用いて決定している。
単離
| 単離 | Actinoplanes sp. ATCC 33076 (2015) |
|---|---|
| 生物活性 | several cancer cell lines displayed significant potency (glioblastoma: LD50 = 5.6 × 10−5 μg/mL) |
| 構造決定 | ESI-MS, 1H NMR, 13C NMR, COSY, HMBC, ROSEY, UV |
| 構造的特徴 | Complex polycyclic macrolide 19 asymmetric centers |
逆合成解析

全文和訳
Neaumycinの最初の複合体は、2012年にShenらによって土壌放線菌Streptomyces sp. NEAU-x211から単離された1)。その後、Neaumycinの構造は2015年に大幅に改訂され、立体化学はないものの、Neaumycin Aと複合体Neaumycin Bが単離された2)。2018年、JensonとFenicalらは、バハマ諸島で採取された熱帯性褐藻Stypopodium zonaleの表面から、海洋微生物Micromonospora sp.(CNY-010株)から物質を単離した3)。ゲノムデータと二次元NMR研究の組み合わせにより、neaumycin Bは 1 であると推定された。いくつかの癌細胞株に対するNeaumycin Bの予備的in vitro試験では、有意な効力(LD50:5.6×10-5 μg/mL)を示し、特に、最も悪性度の高いタイプの神経膠腫の一つであるU87ヒト神経膠芽腫に対して選択性を示した4)。Neaumycin Bの生理活性は、ドラッグデザインのリード構造として有望である。したがって、Neaumycin B (1)の全合成を開発することは重要であり、大きな関心を集めている5a-c)。ここでは、報告されている構造のneaumycin B (1) の最初の全合成を報告する。
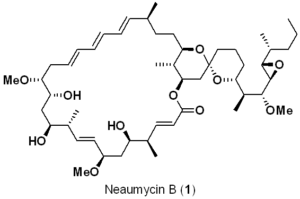
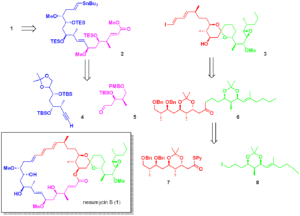
逆合成の観点から(Scheme1)、Neaumycin B(1)を直鎖状の南部(C1-C17)(2)とスピロ環状の北部(C18-C41)(3)に分解し、これらをスティルカップリング(6)とマクロラクトン化により結合させ、脱保護後にNeaumycin B(1)を完成させることを想定した。
直鎖状の南部 2(Scheme 1)は、南西部(7)と南東部(8)のフラグメントの非対称1,2-付加から生じると想定され、一方、北部(3)のスピロケタールコアは、直鎖状のケトン前駆体 4(Scheme 1)から構築され、それが北西部(5)と北東部(6)のフラグメントに分解される可能性がある。
我々の合成は、Neaumycin BのC21-C29セグメントである北西部 5(Scheme 2)から始まった。エポキシド (+)-9(既知の化合物から2段階で調製; これは、ベンジル保護、トリメチルシリルエーテルの加水分解、Cu/TEMPO触媒によるアルデヒド 12 への好気的酸化(8)を経て、カルボニルのα位で硫黄の酸化やエピメリゼーションを起こすことなく(X線参照)、うれしいことに、付加体 11 を得た。酢酸メチルから誘導されたエノラートと 12 のフェルキン-アン選択的アルドール反応により、ジアステレオ選択性(15:1)よく、目的の合成付加体 13 が得られた。ジチオアセタール加水分解9)およびEvans-Saksena還元10)に続く1,3-ジオール保護により、所望の反配置を持つ化合物 15 が得られた。メチルエステルの鹸化とそれに続くチオエステル化により、デカグラムスケールで北西部 5 の合成が完了した。
次に、北東(C30-C41)部 6 に着目した(Scheme 3)。エポキシアルコール (-)-16 (91% ee, (E)-2-hexene-1-ol から Sharpless エポキシ化11)で容易に調製可能) をトリメチルアルミニウム12)で位置選択的に開環すると、粗 1H NMR (>20: 粗 1H NMR (Figure S27 参照) で示されるように、1,2-ジオール 17 (1,2- ジオール/1,3-ジオール) が得られ、二相過ヨウ素酸開裂およびジブロモ-オレフィン化13)により、38 g のジブロモ-アルケン 18 が得られた。18 をn-BuLiに曝露し、ホルムアルデヒドでリチウムアルキニリドを捕捉すると、プロパルギルアルコール 19 が得られた。ルテニウム触媒14a-d)を用いたヒドロキシル基によるトランス-ヒドロシリル化反応により、良好な位置選択性、Z/E選択性、収率でアルコール 20 が得られた。得られたアリルヒドロキシルをMnO2で酸化すると、定量的にアルデヒド 21 が得られ、このアルデヒド 21 は 22 とEvansアルドール反応15)を起こして 23 を形成し、続くトランスアミド化により、所望のsyn配置を持つワインレブアミド 24 が得られた(82%、2工程)。その後、臭化アリルマグネシウムのアミド 24 へのモノ付加反応によりβ-ヒドロキシルケトンが得られ、続いて奈良坂-Prasad還元16a-c) (Et2BOMe, NaBH4)とアセタール保護により 25 が得られた。ヒドロホウ素化/酸化、次いでアッペル反応17)により、北東部 6 が完成した。
北西部と北東部のフラグメント 5 と 6 を手に入れた我々は、それらの結合に適した方法を検討することにした(Scheme 4)。有機リチウム化学もグリニャール化学も、満足のいく収率で目的のカップリングケトンを得ることには成功しなかったが、最近開発されたニッケル触媒を用いた還元的クロスカップリング・プロトコール18)によってフラグメントカップリングが達成され、ケトン 4 がグラム・スケールで76%という良好な収率で得られた。次に、ケトン 4 をメタノール中でp-トルエンスルホン酸にさらすと、脱保護/スピロケタール化が達成され、26 とエピマー 26′ が1:0.8の混合物として得られ、これらはクロマトグラフィーで分離可能であった。嬉しいことに、純粋な望ましくないエピマー 26′ を同じ酸性条件にさらすと平衡が再確立され、それにより、クロマトグラフィー分離/再平衡化のたびに 26 を採取することができ、26 を全体収率67%で得ることができた。H-33とH-28の間の重要なNOE相関(Scheme4, FigureS53参照)により、26 の立体異性が確認された。
C27-OHの3,4-ジメトキシベンジル保護(Scheme 5)を達成するために、26 をまず触媒テトラ-n-ブチルアンモニウムフルオリド(TBAF)19)で処理し、C35-C37を横断する5員シロキサン環の閉鎖に影響を与え、選択的なDMB保護のためにC27-OHを露出させた(詳細はSI参照)。その後、シロキサンをメチルリチウムにさらすと、C35-OHの保護が外され、27 が得られた。
このようにして 27 のC35-C36結合のコンフォメーションがロックされた状態で、あらかじめ導入されたC37シリル基によるA1,3ひずみにより14)、C36-C37オレフィンのエポキシ化がバナジル触媒20)を用いて排他的な合成選択性で進行し、28 が得られた。TBAF21)で脱シリル化し、C35-OHをメチル化すると 30 が得られ、続いてC21およびC23ベンジルエーテルを水素化分解するとジオール 31 が得られた。次にC21一級アルコールを化学選択的に酸化するとβ-ヒドロキシアルデヒド 32 が得られ、これをWittigホスホラニリデン試薬で処理すると22a,b)、優れたE選択性でオレフィン 33 が得られた。うれしいことに、化合物 33 は結晶性であったため、X線結晶構造解析によってC20-C41セグメントにわたる立体構造を明確に確認することができた(Scheme5参照)。
次に、33 のC23-OHのトリエチルシリル保護、エステルの還元、MnO2酸化によりエナール 34 が得られ、これを2,3-ジクロロ-5,6-ジシアノ-1,4-ベンゾキノン(DDQ)で処理してC27-OHの脱保護を行い 35 を得た。最後に、ジヨード(トリブチルスタニル)メタンを用いたアルデヒド 35 の高井-内本オレフィン化反応23a,b)により、収率よく 3a が得られたが、(E,E)/(Z,E)異性体の4:1混合物として受け入れられなかった。
そこで合成経路を見直し、第二世代の北半球(3b、Scheme6参照)を合成した。β-ヒドロキシアルデヒド 32 をまずトリエチルシリルで保護し、高井・内本反応23a,b)を行った。カルボニルに隣接するα-炭素の立体的なバルクにより、CrCl2/Bu3SnCHI2による高井オレフィン化が優れたE-選択性(>20:1)で進行し、対応するビニルスタンナンが得られた23a,b)。その後、ヨウ素化を行い、2段階にわたって収率78%でE-ヨウ化物 37 を得た。3,4-ジメトキシベンジル基のDDQ除去により 38 が得られた。次に、ヨウ化アルケニル 38 とゲルミルスタナン3924a-c)の間のStilleクロスカップリング24c)により、ジエンの異性化を伴わずにジエニルゲルマン 40 が得られた。次に、ゲルマン 40 をN-ヨードスクシンイミドでヨウ素化すると、優れた立体特異性で第二世代の北半球 (3b) が得られた。
直鎖状南西および南東フラグメント(Scheme 7a, b)に目を向けると、アルキン 7 の合成は、既知のアルデヒド 41(D-キシロースから5段階で調製(25a,b))から始まった。マーシャル不斉プロパルギル化26)により、反構成のホモプロパルギルアルコール 42 が収率よく得られたが、ジアステレオ選択性は4:1と控えめであった(Scheme7)。次に、付加体 42 をTBAFで処理し、クロマトグラフィー後に優れた純度のジオール 43 を得た。7 の立体構造を証明するために、ジオール 43 をPMPアセタール化すると結晶 44 が得られ、X線結晶構造解析の結果、南西セグメント(C8-C14)の構造が確認された。その後、シリルエーテル保護により南西フラグメント(7)が完成した。
南東フラグメント(8, Scheme 7b)の合成はβ-ケトエステル 45 から始まった。tert-ブチルジメチルシリル(TBS)保護およびジイソブチルアルミニウム(DIBAL)によるエステルの還元により化合物 47 が得られ、次いで4-メトキシベンジル(PMB)による第一級アルコールの保護後、温和な塩基性条件下でTMS基を除去すると、末端アルキン 48 が得られた。半水素化、水素化ホウ素化/酸化、Cu/(2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO)触媒による好気的酸化8)を経て、Southeasternフラグメント 8 が完成した。
アルキン 7 とアルデヒド 8(Scheme8)をマルチグラムスケールで調製した後、Carreira不斉アルキニル化28)を行うと、優れた収率とジアステレオ選択性で付加体 51 が得られた。しかし、従来の水素化アルミニウム試薬(LiAlH4、Red-Al)を用いた三重結合のトランス還元では、所望のアリルアルコールを高い収率で得ることはできなかった。そこで我々は、Ru触媒を用いたヒドロスタネーション/デスタネーション法29a,b)に着目した。53 のC7-ヒドロキシルの立体化学的評価は、Mosherエステル分析30)によって達成された(Table S27参照)。次にメチル化により化合物 54 が得られ、50%トリフルオロ酢酸(TFA)水溶液にさらすと、主要生成物としてトリオール 55 がきれいに得られた。選択的なトシル化およびエポキシド閉環、次いでシリル保護基の再置換により、高収率(92%、3段階)で 56 が得られ、これをリチウムトリメチルシリル(TMS)アセチリドによるBF3アシスト求核開環にかけると、ほぼ定量的収率でアルコール 57 が得られた。Meerweinの試薬を用いたメチル化により 58 が得られ、続いてTBAFでグローバルなシリル基を除去し、ヒドロキシ基をトリエチルシリルで再保護した。次に 59 のPMB保護基を除去し、得られたアルコールのDess-Martin酸化31)に続いてHorner-Wadsworth-Emmons(HWE)オレフィン化32)により 61 を得た。最後に、末端アルキンの臭素化とヒドロスタン化33)により、位置選択的および立体選択的にビニルスタンナンが形成され、1.26 gスケールで直鎖状南半球 2 の合成が完了した。
北半球 (3b) と南半球 (2) の両方の調製に成功し(Scheme9)、Stille結合反応6)で 2 と 3 を結合させた。嬉しいことに、目的の(E,E,E)-1,3,5-トリエン 62 は、二重結合の異性化もなく、200 mgのスケールで素晴らしい収率で生成した!次に、このメチルエステルを加水分解し、水酸化トリメチルスズによるトランスエステル化を経て、セコ酸 63 を得た34)。向山の条件を用いたマクロラクトン化35)により、シリルで保護された大環状化合物 64 が良好な収率で得られた36)。最後に、トリエチルシリル基のグローバルな除去は、温和な条件(TBAF、HOAc、0℃)できれいに進行し、白色粉末として単離されたNeaumycin B(1)の報告された構造の合成を完了した。注目すべきは、1 が90 mgのスケールでシングルバッチで調製され、全体の収率が2.3%であったことである。
残念なことに、合成した 1 の1H NMRスペクトルは、Fenicalら3)が報告したスペクトルと大きく異なっていた。重要なことに、合成 1 の立体化学的帰属は、各フラグメントのX線結晶構造解析から得られた(Figure1)。すなわち、化合物 33、44 および 50(詳細はSI参照)の結晶構造から、合成Neaumycin BのC20-C41、C8-C14およびC3-C6にまたがる立体化学がそれぞれ確認された(Figure 1参照)。C7における立体異性は、Mosherエステル分析によっても確認された(詳細はTable S27参照)。ここで報告された証拠に基づき、ここで調製された合成Neaumycin B (1) は、Fenicalら3)によって報告された構造と一致すると確信する。
スキーム
化合物番号は全文和訳と異なります
北西部 (5) の合成
9 → 10 + 11
エポキシドの開裂
10 → 12
1,2-ジオール選択的トシル化
12 → 13
エポキシ化
14 → 15
イミデートを用いたPMB保護
15 → 16
DIBAL還元
16 → 17
ジチアン
17 → 18
シリル付加
18 → 19
ブルック転位に続く 13 の付加
19 → 20
ベンジル保護
20 → 21
銅触媒を用いたTEMPO酸化
21 → 22
アルドール反応
22 → 23
ジチアンからケトンへの変換
23 → 24
エバンス・サクセナ還元
24 → 26
ジメチルアセタールによるジオールの保護
26 → 5
加水分解に続くチオエステル化
北東部 (6) の合成
28 → 29
29 → 30
30 → 31
31 → 32
32 → 33
33 → 35
エバンスアルドール反応
35 → 36
36 → 37
37 → 38
奈良坂・プラサード還元
38 → 39
39 → 40
40 → 6
アッペル反応
第一世代北部 (3a) の合成
5 + 6 → 4
4 → 41 + 41′
41 → 42
42 → 43
43 → 44
44 → 45
45 → 46
46 → 47
47 → 48
48 → 50
50 → 51
51 → 52
52 → 53
53 → 54
54 → 55
55 → 56
56 → 57
57 → 58
58 → 3a
高井・内本オレフィン合成
第二世代北部 (3b) の合成
53 → 59
59 → 60
高井・内本オレフィン合成
60 → 61
61 → 62
62 → 64
64 → 3b
南西部 (7) の合成
65 → 67
マーシャル プロパルギル化
67 → 68
68 → 7
68 → 69
南東部 (8) の合成
70 → 71
71 → 72
72 → 73
73 → 74
74 → 76
76 → 77
77 → 78
78 → 8
南部 (2) の合成
7 + 8 → 81
カレイラ アルキニル化
81 → 82 + 82′
82 + 82′ → 83
83 → 84
84 → 85
85 → 86
86 → 87
87 → 88
88 → 89
89 → 90
90 → 91
91 → 92
92 → 93
93 → 94
デス・マーチン酸化
ホーナー・ワズワーズ・エモンズ反応(HWE反応)
94 → 2
Neaumycin B (1) の全合成
2 + 3b → 95
95 → 96
96 → 98
98 → 1
参考文献
1) Streptomyces sp. NEAU-x211から単離されたNeaumycinの単離文献。現在とは大きく異なる構造が示されている。
2) Neaumycinの構造が本文献で紹介した平面構造に改訂された。
3) ゲノムデータと二次元NMRの結果から、Neaumycinの立体構造が示された。
4) 神経膠腫(グリオーマ)に関する文献
5) 過去に行われたNeaumycin Bの部分合成 a) 南部セグメントの立体選択的な合成 b) C1-C18部の合成 c) C1-19部、C23-C35部の合成
6) John K. Stilleによるスティルカップリングの報告
7) a)ジチアン(硫黄原子を2つ有する飽和6員環)を用いた、複雑な天然物骨格の構築 b) アニオンリレーケミストリー(ARC)を用いたRhizopodinの部分合成 c) ARCを用いた複雑な天然物骨格構築の歴史
8) Cu/TEMPOでの酸化
9) 脱チオアセチル
10) a) Me4NHB(OAc)3を用いた、β-ヒドロキシケトンの立体選択的な還元 b) サクセナ・エバンス還元の報告
11) 立体選択的なエポキシ化
12) エポキシドの位置選択的な開裂
13) アルデヒドからアルキンを得る手法
14) a) トリメチルシリル(TMS)がアリルアルコールのエポキシ化における立体選択性に与える影響 b) アルキンに対するルテニウム触媒を用いた位置選択的なヒドロシリル化によるヒドロキシケトンの合成 c) アルキンを利用したSoraphen Aの全合成 d) アルキンを利用した天然物合成の歴史
15) エバンスアルドール反応の報告
16) a) β-ヒドロキシケトンから1,3-ジオールへの立体選択的な還元 b) β-ヒドロキシケトンから1,3-ジオールへのsyn選択的な還元 c) ボランを用いた、β-ヒドロキシケトンから1,3-ジオールへのsyn選択的な還元
17) アッペル反応の報告
18) Zr/Niを用いたワンポットでのケトン合成
19) アルキニルベンジルジメチルシランから環状アルケニルジメチルシロキサンを得る手法
20)
21)
22)
23)
24)
25)
26)
27)
28)
29)
30)
31)
32)
33)
34)
35)
36)